Additional data tables and analysis not included in the main 2025 Annual SHOT Report.
Additional case studies
Patient death
Case 18.6: Inappropriate delay in provision FFP possibly contributes to patient death
A patient was admitted to the ED after suffering a hypovolemic cardiac arrest due to trauma. The MHP was activated. The policy in this hospital was for FFP requirements to be communicated by clinical staff upon MHP activation, but this did not occur. As such, no FFP was thawed by the BMS and the availability of pre-thawed FFP was not checked by the BMS or queried by the clinical team. The patient received seven units of red cells and three units of FFP. There was a delay in obtaining haemostatic control and the patient passed away.
The BMS was a new trainee BMS with minimal experience of MHP activations. They were working alone during a night shift, covering haematology and transfusion departments. The report noted challenges with workload and staff provision, and gaps in communication. The policy in this hospital has been updated to automatically thaw FFP upon MHP activation. Staff were also reminded about the importance of clear communication and the BMS was asked to perform a reflective practice. There were no actions shared regarding staffing provision, workload or training for newly qualified members of staff prior to lone working.
Major morbidity
Case 18.7: Major obstetric haemorrhage – laboratory delay in emergency blood provision causing major morbidity
A woman was experiencing a major antepartum haemorrhage and was immediately taken for Emergency caesarean section under general anaesthesia. The anaesthetist called the laboratories emergency telephone to activate the MHP but there was no answer. They tried a further two times, but the call remained unanswered causing a sense of panic for theatre staff. They then called the other enquiry call numbers for the laboratory with no success. Advice was sought from switchboard to obtain a bleep number and finally got to speak to the BMS on duty.
The BMS was lone working overnight and currently taking a break. It took 10-15 minutes to contact the laboratory whilst the patient was experiencing acute bleeding, which would become life threatening without immediate intervention. Due to the urgency for blood at this point, there was insufficient time to issue any group specific units to the patient on the LIMS, resulting in emergency group O D-negative units being taken. The units were taken immediately and transfused and the patient stabilised.
On investigation the laboratory has a mobile phone as well as a landline phone that simultaneously ring when a MHP call is activated. On this occasion, the BMS accidentally left the mobile phone in the laboratory when they went to the cafeteria for a short break. There was also further confusion caused as switchboard had an outdated number listed to activate the MHP.
Following the event, laboratory staff were reminded that they must hand over the mobile phone during the shift handover and must carry it with them at all the times while on duty. Switchboard have also updated their contacts.
Case 18.8: Woman sensitised to the D antigen in the context of many systemic issues
A female patient of childbearing potential, who was O D‑negative, was inadvertently transfused with O D‑positive blood during an emergency theatre request when no valid pre‑operative sample was available. An initial group‑and‑screen sample had been rejected due to labelling errors. The sample rejection was communicated to the department who requested the group and screen, but this information was not passed on to the admitting ward. The lack of a valid result was not noted by the clinical team for the next 2 days, resulting in the patient going to theatre without appropriate blood cover 48 hours later. On later admissions, the patient was found to have developed anti‑D, anti-C and anti‑E antibodies, confirming she had been sensitised by the incompatible transfusion.
Several contributing factors were identified. The laboratory was operating severely below its planned staffing levels because senior personnel were away on intensive LIMS training, leaving remote supervision in place and only one qualified BMS present, when expected provision was three. The BMS on duty was competent but also supervising a trainee and reportedly dealing with personal health concerns. National shortages of O‑negative blood meant staff were consciously trying to preserve stock, and the incident occurred just one day before a formal amber alert was declared. Additionally, the LIMS does not generate D compatibility alerts during emergency issue, and the designated colour‑coded emergency units were not used, using them might have visually prompted recognition of the mismatch.
Communication gaps between clinical teams and the laboratory, the use of non‑emergency stock instead of dedicated emergency units, and the lack of complete documentation around the emergency request all contributed to the incident. This event occurred within a context of system limitations, workload pressure, inappropriate staffing and suboptimal communication.
Case 18.9: K-positive blood issued to woman of childbearing potential causing sensitisation to the K antigen
In 2023, two K‑positive red cell units were issued and transfused to a female patient of childbearing age. The patient subsequently developed anti‑K, identified during laboratory testing in 2025, indicating sensitisation caused by the earlier transfusion.
The incident investigation found no evidence explaining why K‑positive units were selected due to the time elapsed. The units were not nearing expiry, ruling out stock‑management pressures. Contributing factors identified by the reporter included inadequate checklist design, lack of appropriate LIMS functionality to alert staff when a K‑positive unit is selected for a woman of child‑bearing potential, knowledge gaps and likely inattention to detail during the issue process, though these are hard to determine due to the time that has elapsed since the event.
Case 18.10: Inadvertent sensitisation to the K antigen impacts future pregnancies and family planning for a woman of childbearing potential
In 2022, a pregnant woman received two units of red cells following a caesarean section; one of these units was later found to be K‑positive. During antenatal screening in 2024 for a subsequent pregnancy, the patient was identified as having developed anti‑K, indicating sensitisation linked to that earlier transfusion. This was further compounded by incomplete antibody monitoring, as a sample in later pregnancy was not sent onto the reference laboratory for titration. The error was not detected at the time of issue due to limitations in the checklist design, which did not adequately support identification of K incompatibility.
Human factors issues also contributed. The laboratory scientist involved was a trainee who was noted by the reporter to demonstrate overconfidence and a reluctance to seek assistance, which made training challenging and increased the risk of missed details. Possible fatigue was noted, as the staff member had been working additional shifts during a period of low staffing. Although workload on the day was moderate, organisational challenges such as staff shortages and high turnover—already recognised on the Pathology risk register—likely exacerbated these risks. Unclear wording and functionality of LIMS alerts also impacted as corrective actions included implementing a new LIMS with improved warnings and override procedures, plus the addition of specific luggage tags to identify K-positive or untested red cell units. The mother and baby were closely monitored throughout the pregnancy, and the baby was born unaffected, however the mother has chosen to not peruse any future pregnancies due to potential risk to fetuses.
Case 18.11: Patient requiring resuscitation prior to blood component provision due to communication issues
A patient with GI bleeding was in the resuscitation are of the ED at 13:35 and was found to have a low Hb of 39g/L and was demonstrating marked clinical deterioration. There was difficulty obtaining patient samples and the doctor decided to activate the MHP. They called the transfusion laboratory on the designated number to activate the MHP. The BMS who answered the phone asked if a group and screen had been taken. The doctor emphasised that these would be completed as soon as possible but the patient required immediate MHP support. After 5 minutes, the doctor called the laboratory again on the designated number to confirm that the MHP had officially commenced but again the BMS questioned whether a group, screen and crossmatch sample had been taken. Again, it was highlighted to the BMS that the patient was in immediate need of blood components and issuing the components was more important than sending the samples at that moment in time. At 13:58, the patient went into pulseless electrical activity arrest and cardiopulmonary resuscitation (CPR) was started. The patient had already received 2L of IV fluid as stat. At 14:11, two units of emergency O D-negative red cells arrived while CPR was ongoing. The two units were administered and at 14:14 the patient was noted to have returned to spontaneous circulation (ROSC). At 14:35, four units of fresh frozen plasma and four units of group-specific red cells arrived. The patient was stabilised and admitted to the critical care unit a short time later. The delay in the initial transfusion was around 25 minutes. The patient passed away from their underlying condition.
On investigation there were communication failures and misunderstandings between the ED and the transfusion laboratory. The BMS was under the impression that group-specific red cell units were required and was waiting for the group and screen samples to arrive. The laboratory should have recognised the urgency for blood and informed the clinical area that emergency O D-negative units were available to be collected from the laboratory by clinical staff. It appeared that the clinical area staff were not aware of this procedure for emergency red cells. Corrective actions included MHP simulations involving both clinical and laboratory staff and education sessions for clinical staff.
Case 18.12: Delay in intraoperative blood provision leading to cardiac arrest
A patient was admitted for an extensive surgical procedure with a Hb of 137g/L. Two group and screen samples were sent to the laboratory prior to the start of surgery due to potential for bleeding. A member of theatre staff rang the blood transfusion laboratory at 12:37 to request 2 units of crossmatched red blood cells for intraoperative bleeding. The patient’s Hb at this point was 107g/L. A member of laboratory staff began processing this request.
When no blood had been made available an hour later at 13:40, a member of theatre staff rang the laboratory requesting two units of crossmatched red cells and two units of FFP. The laboratory staff started processing this request on a different sample than the initial request. There was no system in place within the laboratory to identify that a crossmatch had been requested and started for the patient on the previous sample. There was no specific urgency for blood provision communicated at this time, though clinical staff had stated that blood loss was 2.5L during the phone call with the laboratory.
At 13:42 another request for crossmatched red cells was made via telephone to the laboratory. The laboratory staff member taking this call advised that fully crossmatched red cells would take at least 40 minutes to prepare. They were unaware that red cells were waiting to be issued from the initial crossmatch request at 12:37. After querying the urgency of the request, the theatre confirmed they wanted group-specific blood.
When the laboratory staff attempted to issue group-specific blood, the LIMS showed a sample mismatch as staff were scanning against the wrong sample and staff were unable to issue the group-specific blood.
At 13:55 the MHP was activated, at 13:58 the porter collected the emergency issued red cells, the crossmatched red cells and FFP. At 14.03 the patients Hb was 40g/L. In total they received nine units of red cells, six of FFP, two of cryoprecipitate and two adult therapeutic doses of platelets. The patient did suffer a cardiac arrest and required admission to the intensive care unit but survived.
On investigation, LIMS had been introduced 2 months previously and this was the first MHP activation since launched. The sample issues were not identified during pre-implementation testing. Communication challenges regarding urgency of the request were reported and the MHP could have been activated in a timelier manner. Corrective actions included testing of the LIMS and communication aids for the theatre teams regarding blood availability.
This case highlights the importance of robust LIMS validation prior to implementation, as unintended outcomes may occur if a situation is overlooked during testing (in this case an unnecessary delay due to lack of LIMS functionality highlighting previous requests for this patient). In addition to this, there should be clear methods of communication for laboratory staff to be able to identify if components have already been requested for a patient.
Furthermore, there should be SOP for taking requests for blood via the telephone, which includes querying the urgency of the request to assist in collaborative communication with the clinical area. Laboratory staff should also have adequate knowledge to identify when requests are urgent. The language of a bleeding patient in theatre, and the blood loss being specified as being 2.5L should have further prompted the laboratory staff to query whether this request was urgent or an emergency. The SHOT laboratory communication toolkit (https://www.shotuk.org/resources/laboratory-communications-toolkit/), published in 2025, contains tools to assist with the various situations which occurred here:
- A telephone request form which includes questions regarding urgency
- A communication guide for clinical areas to assist in understanding the difference in delivery times and components provided for different requests (e.g. crossmatched or emergency issues)
- A handover log which includes ongoing crossmatches
Information technology errors
Table 18.4: Information technology involvement in laboratory errors in 2025 (n=341)
| How was IT involved with the error | Total |
|---|---|
| Lack of functionality/algorithms in the system to support safe practice | 77 |
| IT could have prevented the error | 45 |
| Warning flag in place but not heeded | 38 |
| System not used correctly | 36 |
| Failure to use flags and/or logic rules | 31 |
| Computer or other IT systems failure | 20 |
| Warning flag not updated or disabled | 17 |
| Lack of interfacing/interoperability | 16 |
| Failure to link, merge or reconcile computer records | 16 |
| System not configured correctly | 14 |
| Failure to consult or identify historical recor | 9 |
| Incorrect results entered or accessed manually | 8 |
| Incorrect patient details selected from IT system | 7 |
| Printing error | 1 |
| Other | 6 |
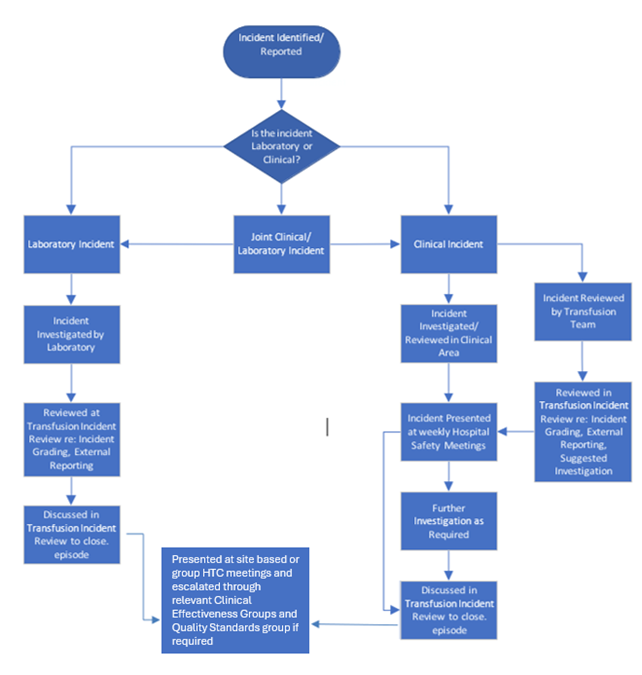
Incident management in transfusion
Author: Peter Baker, SHOT Laboratory Working Expert, Liverpool Clinical Laboratories.
The following is an example of how transfusion incidents can be effectively managed and monitored within a healthcare organisation. SHOT would like to thank Liverpool Clinical Laboratories for sharing their experiences.
Overview
The transfusion incident management system has evolved over many years and now follows a structured, multidisciplinary pathway. All reported transfusion‑related incidents reported via the local incident reporting system—whether clinical or laboratory‑based are automatically emailed to the Hospital Transfusion Committee (HTC) Chair, the consultant responsible for transfusion, transfusion practitioners, the transfusion service manager, laboratory managers, and senior BMS across all four laboratory sites.
Timely review of new incidents is essential to ensure early understanding of risks, initiation of remedial actions, and product recall when appropriate.
Multidisciplinary team (MDT) review process
A dedicated transfusion MDT meets weekly to review all new incidents. The team:
- Confirms the accuracy of the initial risk assessment
- Reviews and approves remedial actions
- Determines whether external reporting is required
- Identifies whether an after‑action review (AAR) is necessary
- Allocates specific actions to responsible clinical or laboratory personnel
- Tracks progress of previously reported incidents
- Ensures that investigations are completed to an appropriate standard
The MDT is also authorised to close incidents once all actions have been completed satisfactorily.
Occasionally, clinical areas close incidents prematurely. When reviewed by the MDT, it may become evident that key contributing factors—particularly human factors—have not been considered. In such cases, the incident is re‑opened and managed through the MDT process to ensure full oversight and governance. The role of the transfusion practitioner is vital in this context, as they liaise with the clinical areas and provide support. In addition, the event will be assigned to the clinical area on the local reporting system, so can be monitored through clinical governance structures that the ward sits within.
Upon completion and closure, all transfusion incidents are escalated through the HTC and the Trust’s wider governance structures.
Figure 18.6: Incident management flow chart
DATIX
The Trust utilises the DATIX incident management system. Historically, DATIX could not be modified, and teams were required to use the standard 5×5 matrix based on actual harm rather than potential harm. Following extensive discussions, the system was successfully adapted to support transfusion‑specific requirements, including assessment based on potential harm. Adoption of PSIRF continues to present challenges, particularly where regulatory reporting still requires a root cause analysis; however, a shared understanding has now been reached regarding how the two approaches can be aligned.
Integrated transfusion risk assessment
In addition to standard reporter and incident details, DATIX has been customised to embed the Quality Risk Assessment (QRA) Tool, originally developed by the laboratory. This replaces the generic 5×5 matrix with a transfusion‑appropriate 3×3×3 matrix incorporating:
- Likelihood
- Impact (based on potential harm)
- Detectability
A secondary QRA section is provided for incidents that remain open beyond their target completion date.
Categorisation of contributory factors
DATIX now includes specific categories for investigation findings, including:
- Externally reportable
- Equipment/technology
- External environment
- Internal environment
- Organisation
- People
- Processes/tasks
Each category includes drop‑down sub‑options. For example, the People category includes: competence, health, knowledge base, mistake, skill mix, skill base, slip, and teamwork. This enables more accurate tracking and trend analysis of causal factors.
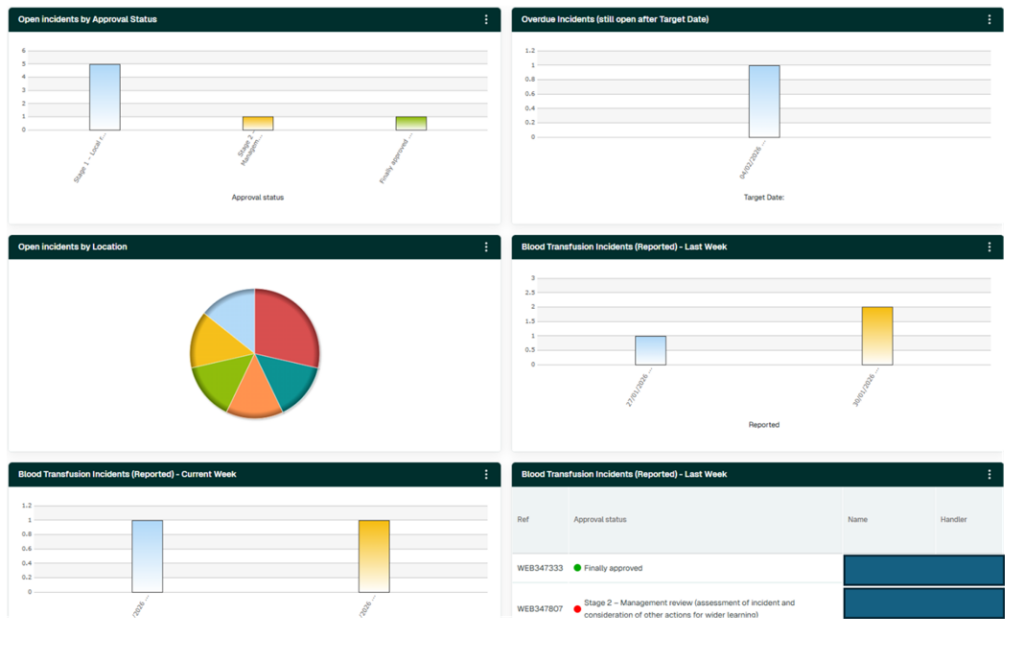
Incident dashboard
A bespoke dashboard supports effective oversight and management. Key features include:
- Clear visualisation of all open incidents
- Highlighting of incidents exceeding target dates
- A pie chart showing distribution of incidents by site/area
- Summary boxes showing incidents reported this week and last week
- A dedicated section listing outstanding actions
All dashboard elements are interactive, allowing direct navigation to relevant incident groups. Incidents exceeding target dates are clearly flagged, prompting reassessment of risk to ensure current mitigation measures remain adequate.
Figure 18.7: Example dashboards for incident management
Quality risk assessment tool
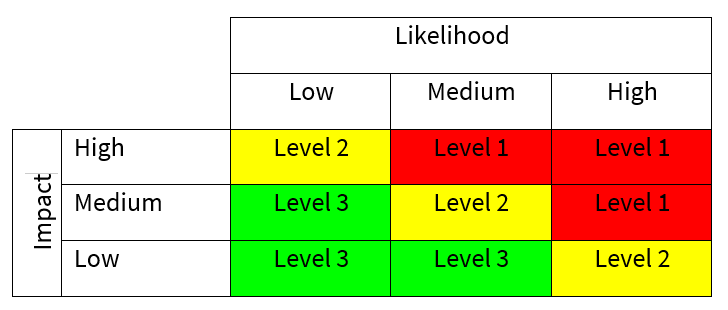
The risk analysis requires the identification of associated risks, along with the criteria defined below, to determine the appropriate measures to mitigate the identified risks on the risk analysis table.
Likelihood is defined as the probability that the event will occur:
| Description | Frequency |
|---|---|
| Low | Unlikely to occur or recur |
| Medium | Possible, but not expected to occur or recur |
| High | Likely to occur or recur, potentially frequently |
Impact reflects the potential effect on patient outcomes, product integrity, and service quality:
- Low: Minor negative impact without patient harm (e.g., small delays, minor product loss, isolated quality standard failure). Could lead to single failure to meet quality standards.
- Medium: Moderate negative impact with potential to affect clinical outcome transiently (e.g., delay to treatment, moderate product loss, repeated failures to meet standards). The impact could have short to medium-term detrimental effect.
- High: Significant potential to adversely affect patient outcome (e.g., missed treatment window, major product loss, major quality standard or licensing failure; potential long‑term consequences). The impact could have significant long-term effect and potentially catastrophic effect.
RISK CLASSIFICATION – Both likelihood and impact can be represented by a matrix for evaluating risk action levels:
DETECTABILITY – The purpose of this stage is to identify if the risk event can be recognised or detected by other means in the system. This might include:
- Preventative controls – which are actions taken to prevent specific undesirable events where impact is likely to be significant (e.g. segregation of duties, access controls, supervision, password checks etc.)
- Detective controls where actions taken to detect or correct undesirable events when they materialise (e.g. monitoring, testing, reconciliation etc.)
- Directive controls which are designed to direct people to take action to achieve a specific outcome (e.g. prescriptive in terms of policies, standards, procedures, training etc.). The probability of risk being detected can be estimated as follows:
| Low | Medium | High |
| Detection of error/event is unlikely | Detection of error/event is reasonably likely | Detection of error/event is highly likely |
RISK PRIORITY LEVEL – By combining risk classification with the probability of detection, it is possible to prioritise the fault conditions associated with each problem based upon those areas of greatest vulnerability and to prioritise the risk mitigation measures accordingly.
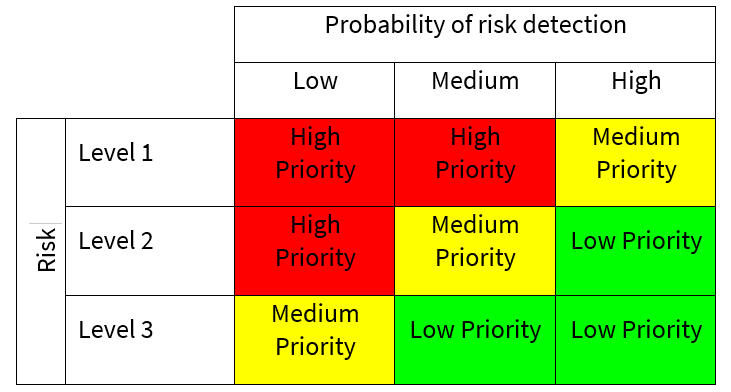
- Low priority: Routine procedural controls or straightforward corrective actions
- Medium priority: Additional testing or enhanced control measures required
- High priority: Comprehensive interventions needed, including strengthened controls to increase detectability or reduce likelihood
Summary
Effective transfusion incident management requires a well‑structured, multi‑professional approach supported by technology and risk stratification tools. A knowledgeable MDT with meeting weekly, ensures:
- Robust and consistent investigation
- Appropriate risk assessment based on potential harm
- Timely implementation of mitigation measures
- Learning from incidents
- System improvements that reduce likelihood or increase detectability
This structured oversight is essential for patient safety, regulatory compliance, and continual improvement of transfusion practice.