Authors: Chris Robbie and Mike Dawe
Link to PDF chapterKey SHOT messages
Key findings
- Apart from a reduction in the number of serious adverse events (SAE) assessed in the reporting year, there is very little change in reporting patterns.
- Staffing and workload factors may be evident in up to 73.5% of all human and system-related SAE.
- The quality of reports and investigations frequently does not meet the expected requirements of the Good Practice Guide (GPG) (EDQM, 2025).
Gaps identified
- Investigations are not conducted according to the regulatory requirements of the GPG.
- Initial reports fail to identify the preventable causes of error.
- Corrective measures do not initially address the human factors involved.
- Many reports still focus on staff for errors and overlook the weaknesses of processes, procedures and systems missing the opportunity for improvement.
Good practice
- Many investigations and reports are improved following review, assessment and guidance from MHRA.
- Thorough investigation has identified weaknesses and improvements to systems.
Next steps
- Review your Incident management process and procedures against the requirements of the GPG (Chapter 9).
- Improve the quality of investigations and reports to reduce the number of requests for further information.
Glossary, acknowledgments and reference list
Please access these links for the Glossary for all abbreviations, Acknowledgments and References used.
SABRE report data
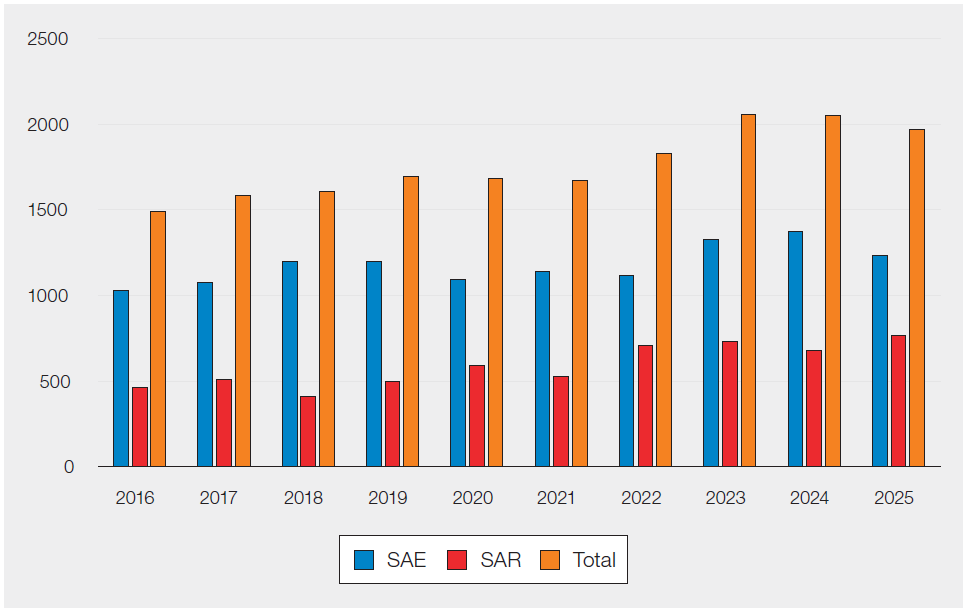
Table 30.1 and Figure 30.1 show the total numbers of reports and the numbers of reports submitted as SAE and serious adverse reactions (SAR) for the previous 10 years.
Table 30.1: Submitted confirmation reports 2016–2025
| Category | 2016 | 2017 | 2018 | 2019 | 2020 | 2021 | 2022 | 2023 | 2024 | 2025 |
|---|---|---|---|---|---|---|---|---|---|---|
| SAE | 1027 | 1076 | 1198 | 1197 | 1093 | 1143 | 1118 | 1325 | 1371 | 1215 |
| SAR | 464 | 508 | 408 | 497 | 590 | 526 | 710 | 731 | 677 | 746 |
| Total | 1491 | 1584 | 1606 | 1694 | 1683 | 1669 | 1828 | 2056 | 2048 | 1961 |
Figure 30.1: Submitted confirmation reports 2016–2025
Serious adverse events n=1215 (-156)
Definition (Department of Health, 2005)
Any untoward occurrence associated with the collection, testing, processing, storage and distribution, of blood or blood components that might lead to death or life-threatening, disabling or incapacitating conditions for patients, or which results in, or prolongs, hospitalisation or morbidity.
Table 30.2 Total number of SAE reports by event category
| Event category | Number of reports |
|---|---|
| Materials | 1 |
| Apheresis collection | 2 |
| Testing of donations | 5 |
| Processing | 5 |
| Whole blood collection | 8 |
| Distribution | 15 |
| Donor selection | 83 |
| Storage | 271 |
| Other | 825 |
| Grand total | 1215 |
Table 30.2 shows the total number of SAE reports received by event category. Proportions of reports received remain similar to previous years although it is noted that there is a marked reduction in those classified as ‘other’.
Storage data n=271 (-21)
Storage remains the second largest individual error category (after ‘other’) and comprises of all BSQR reportable storage SAE in both the laboratory and clinical areas. The MHRA Haemovigilance Team lead has broken this category down further to try and identify specific storage error sub-types, Table 30.3. For a description of the subcategories used, see Appendix 1.
Table 30.3: SAE storage error sub-classifications
| Storage sub-classification | Total |
|---|---|
| Incorrect storage of component | 134 |
| Component expiry | 38 |
| Sample expiry | 33 |
| Return to stock error | 28 |
| Failure to action alarm | 14 |
| Storage temperature deviation | 10 |
| Security | 8 |
| Miscellaneous | 5 |
| 30- or 60-minute rule | 1 |
| Total | 271 |
Other n=825 (-131)
Table 30.4 shows the number of reports in the ‘other’ category of SAE. There has been a decrease in events that fall into this category. It is difficult to know the exact reasons for this, but it could be due to the following:
- Improvements to systems, processes and procedures due to corrective and preventive action (CAPA)
- Recovery to ‘normal’ levels of reporting following the 2024 South London cyber-attacks
- Delays in completion of reports
- Unknown changes in reporting practices
- Other factors
Please see Appendix 2 for a description of the subcategories.
Table 30.4: Other subcategory by type
| ‘Other’ subcategory | Total |
|---|---|
| IBCI – incorrect blood component issued | 188 |
| PTTE – pre-transfusion testing error | 171 |
| SPE – sample processing error | 146 |
| CCE – component collection error | 118 |
| CLE – component labelling error | 75 |
| DEE – data entry error | 72 |
| FR – failed recall | 19 |
| CATPD – component available for transfusion past de-reservation | 9 |
| IBCO – incorrect blood component ordered | 9 |
| ECAT – expired component available for transfusion | 8 |
| UNSPEC – unspecified | 7 |
| HD – handling damage | 2 |
| IBCA – incorrect blood component accepted | 1 |
| Total | 825 |
Human and system error categories and human factors
The MHRA assign a category on review of an SAE report to reflect the most prominent causative factor. Assessment of these reports can distinguish between events caused by system errors and human errors (slips/ lapses/omissions). For a description of the categories used, see Appendix 3.
The MHRA Haemovigilance team thoroughly assess the reports received to understand the human factors and causative factors that affect each reported SAE to ensure that the incidents are not simply attributed to be due to human error. Human error assumes that the staff involved are solely responsible for the error without assessing the quality management system (QMS) for weaknesses and improvements. Whether the word ‘blame’ is used or not, failure to investigate adequately and assigning reports as human error without justification will overlook potential improvements to systems, process, and procedures. It increases the risk of harm to patients through repeat errors and affects staff psychological safety, morale and well-being. It is for these reasons, the MHRA will continue to request further information if the submitted reports do not meet the minimum requirements of the BSQR and GPG. For further discussion, see section below, ‘The quality of incident investigation and haemovigilance reporting’.
Figure 30.2: Human/system error subcategories (n=1198)
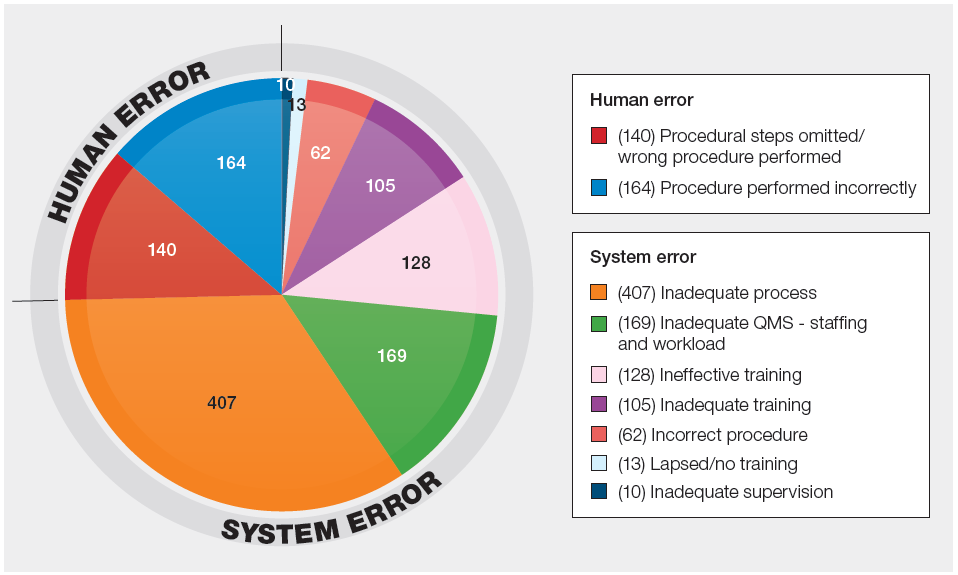
NOTE: These numbers should be used as guidance only. The quality of this data is limited by several factors.
- The incidents are usually the result of many causal and contributory factors. The subcategory chosen reflects the most likely reason for the main SAE category. If multiple factors are involved relating to the QMS, then ‘inadequate process’ has been chosen as the subcategory rather than choosing a category that best fits the main SAE reported.
- The subcategory chosen is based on the information in the report. A limited investigation or a report which does not provide the MHRA with enough information to potentially subcategorise appropriately.
The proportions of reports that fall into these subcategories remain broadly similar to previous years, but there are some notable differences.
- While the proportion of reports assigned as ‘inadequate QMS – staffing and workload’ has dropped slightly from last year (14.1% from 15.4%), it is now the second most frequent category (this was the third most frequent category in 2024).
- The proportion of reports assigned as ‘inadequate process’ has increased from 30.9% to 34.0%.
- Since ‘inadequate process’ not only describes a report with a single weakness identified within a process, but it also describes a report with multiple identified system, process or procedural weaknesses involved.
- Slips, lapses and omissions account for 25.4% of reports (from 27.9% previously)
- This does not necessarily mean that 25.4% of reports are due to human error. It simply means the report has not identified any system issues that need to be addressed.
- These reports will either have been assessed as such because they ‘justify’ human error as a root cause, or because the investigation has not identified any improvements to any weak areas of the QMS.
- When you account for reports which specify staffing and workload issues, reports assessed as ‘inadequate process’ (for multiple causes) and ‘human error’ (due to sub-optimal investigations) the true figure for SAEs relating to staffing and workload could be between 14.1% and 73.5%.
Serious adverse reactions n=746
Definition (Department of Health, 2005)
An unintended response in a donor or in a patient that is associated with the collection, or transfusion of blood or blood components that is fatal, life-threatening, disabling or incapacitating, or which results in or prolongs hospitalisation or morbidity…blood establishments and the person responsible for the management of a hospital blood bank shall notify the Secretary of State (Competent Authority) of any serious adverse reactions observed during or after transfusion which may be attributable to the quality or safety of blood or blood components:
(i) Collected, tested, processed, stored or distributed by the blood establishment, or
(ii) Issued for transfusion by the hospital blood bank
Summary of SAR report data
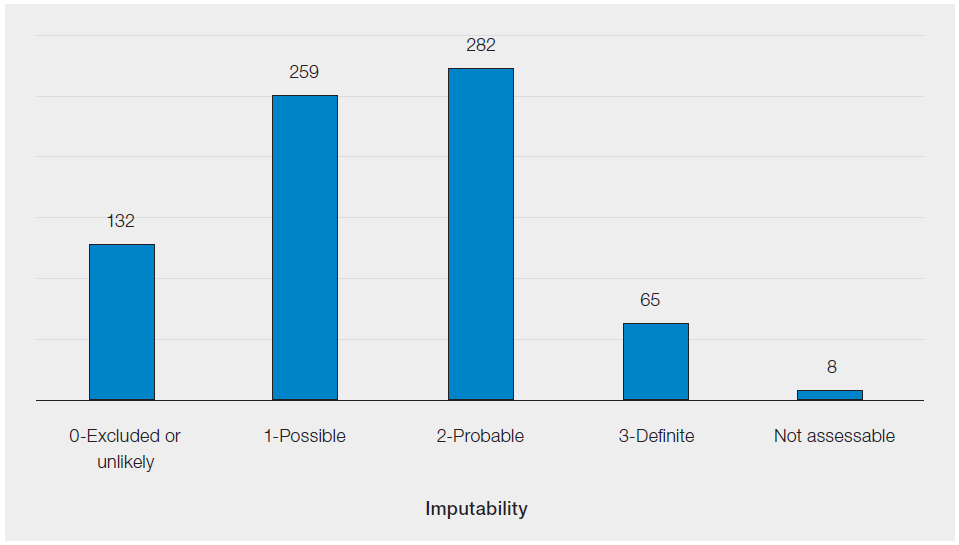
Only the total SAR figures that qualify for reporting to MHRA under the BSQR are provided in this chapter, see Figure 30.3.
Figure 30.3: SAR reports, by imputability, reported to SABRE in 2025 (n=746)
Inspection report
An overview of the compliance management escalation processes used by the good manufacturing practice (GMP) inspectorate, including information on the Inspection Action Group (IAG) and Compliance Management Team (CMT) referral processes, is available from the MHRA inspectorate blog: https://mhrainspectorate.blog.gov.uk/2017/02/06/overview-of-compliance-management-escalationprocesses-used-by-the-gmp-inspectorate/ (Moon, 2017).
Summary of significant issues identified at inspected sites has remained fairly consistent from the previous year and are listed here.
Management of change
The control of change continues to be a deficiency that is commonly raised at blood inspections. The deficiencies raised include:
- The absence of a user requirement specification.
- This must contain all relevant essential and desirable aspects of what a site is intending the change to achieve. For example, the management of data and data integrity between laboratory information management systems (LIMS).
- The lack of a validation master plan to guide management through the validation and qualification of the change.
- This is a management of the validation aspects of the change and shouldn’t be confused with a validation policy.
- Inadequate or absence of a risk assessment and actions to mitigate risks.
- Consider what all of the risks are concentrating on the individual rather than the strategic risks involved.
- The lack of evidence of sign off of stages of the change control prior to implementation.
- This can be electronic signatures i.e. Q-Pulse.
- The lack of validation evidence to show that the system was fit for task before implementation.
- If it’s being validated and approved for implementation, then it needs traceable evidence that supports the decision to implement the change.
- Failure to carry out a post-implementation effectiveness check.
- Include this in the validation master plan.
Management of non-conformances
The management of non-conformances is regularly raised as a deficiency due to the following:
- Inadequate investigation for an appropriate root cause therefore the inadequate implementation of an effective CAPA to avoid reoccurrence.
- For example, reminding staff to follow the correct procedure without identifying why the correct procedure was not followed in the first place.
- CAPA did not address root causes identified by the investigation.
- For example, the investigation states staff were lone working and busy. This may suggest a resourcing issue so have you checked your resourcing plans.
- Failure to consider the potential for harm as well as actual harm, especially healthcare organisations using the Datix system.
- Most incident management systems can be configured to assess potential and not just actual harm.
- The lack of an adequate justification for human error being identified as a root cause.
- The focus has been only on individual/s and not the processes and systems in place and are they adequate and effective.
- Procedures did not ensure that investigations and CAPA would be completed in a timely manner.
- Are you allocating enough resource and time allocated to carry out effective deviation management processes?
- The lack of justification for the late closure of deviations and performing impact risk assessments.
- Review relevant standard operating procedures (SOP) and record a justification for late closure of incidents.
- Tracking and trending systems employed not identifying recurring problems due to an emphasis on consequence rather than root cause.
- For example, where equipment failures are noted, to record what equipment failed and the reasons for failure. Important to also note if these distinct findings were being tracked and trended.
The availability of trained and competent staff
Issues with adequate capacity within the laboratory is an ongoing problem and is often raised as highlighted by:
- The absence of an effective resource management plan or similar document to ensure adequate management of blood transfusion operations and the quality management system. This includes availability of transfusion practitioner support.
- The inadequate management of risk register entries such as reducing the risk score without an appropriate justification.
- If this has been identified at an inspection or accreditation event, then it should be recorded on the risk register. Additionally, it is important to note deviations occurring due to inadequate resource i.e., whilst lone working.
- Risk scores being reduced before the suggested mitigation was in place and deemed effective.
- Reducing a risk score before effective implementation and evidence of impact. Premature closure should be avoided simply because an additional resource has been identified, the risk may not be truly reduced.
- Staff working significantly above their contracted hours to ensure staff rotas are adequately staffed.
- Additional hours should be recorded and managed to avoid staff burnout.
- Trusts/Health Boards failing to meet several quality metric targets.
- For example, close out targets for deviation management and the completion of self-inspection audits.
Blood collection and training
Blood collection and training was not being adequately managed in that:
- Blood collection training and competency audits showing that organisations were not meeting their key performance indicators for staff blood collection training.
- Please see the comments above regarding resourcing.
- Inadequate systems in place to control infrequent users of the system and blocking staff who had left the organisation.
- Implementation of a stand-alone audit and the use of electronic systems such to block infrequent users.
Computerised patient databases and data integrity measures
There have been several examples where patient and result databases have never been checked for the integrity of the stored data evidenced by:
- There was no record that sites had reviewed the retention of backed-up data on the LIMS.
- The laboratory had no evidence that they had performed any audit checks to detect the potential presence of duplicate patient records on their LIMS database.
- The laboratory had no evidence that the backed-up testing records on the automated blood transfusion analysers had ever been checked.
These can be captured by implementing an audit as part of self-inspection processes and by liaising with local information technology (IT) departments outlining the roles and responsibilities in a document such as a service level agreement.
For further information on MHRA and the regulation of blood please refer to the MHRA website: https://www.gov.uk/topic/medicines-medical-devices-blood/blood-regulation-safety (MHRA, 2023).
MHRA Blood Forum
This was launched in June 2016 as a tool to help those involved in blood component collection, processing, testing and distribution to comply with the EU Blood Directives, UK Statutory Instruments, and good practice requirements. It provides the ideal opportunity for extended communication between peers and allows users to put forward their comments and get ‘real-life’ examples of ways in which they can manage robust quality procedures that ensure compliance and which dovetail with their own business needs and resources.
https://forums.mhra.gov.uk/forumdisplay.php?60-Blood-Forum (MHRA, n.d.).
The quality of incident investigation and haemovigilance reporting
Introduction
The quality of investigations and reports to both MHRA and SHOT are not always in line with the expectations of the regulatory references in Chapter 9 of the GPG. While some are very good or adequate, several reports do not meet the minimum basic standard expected and require follow-up. Each SAE and SAR is a legal document, and may be used, if necessary, in a court of law. These are the reporters’ reports, not the MHRA’s and they should give an adequate account of the error being reported, the root causes and the corrective measures taken to mitigate the causes identified.
Evidence from inspection and SAE review and assessment
Examples of reasons why reports are considered to not meet the regulatory requirements potentially include, but are not limited to:
- Lack of adequate detail in general; some reports are single sentences or even bullet points, some reports only describe what happened and how it happened without identifying why the error occurred.
- The causes identified have not been mitigated with adequate corrective measures.
- Corrective measures are introduced that are not relevant to the causes identified.
- Investigations are focused on the outcomes of errors not detected rather than the causes of the errors made.
- Investigations conclude that staff are at fault rather than investigating the human factors of why staff acted or behaved in the why they did.
- Corrective measures focus on reinforcing measures already in place and have already been shown to have been ineffective otherwise the error would not have potentially occurred.
- A poor understanding of human factors where some reporters still believe these are related to the individual alone.
- Even where human factors are demonstrated to have been understood, corrective measures still target the individuals and not the systems, processes and procedures in use at the time the error was made.
These findings are backed up by evidence from MHRA inspections. The inspection report above lists non-conformances related to investigations and reporting as ‘common findings’ which have remained the same for the last 10-15 years. For example, last year, out of every inspection conducted (n=17) of hospital blood banks one or more non-conformances were raised in this area.
Where deficiencies are noted in a SABRE report the MHRA Haemovigilance team will:
- Assess if the error is likely attributable to simple slips or lapses
- Do a look back for repeated errors and causes
Depending on that review:
- MHRA may close the report and monitor for repeat errors, even it is thought that the genuine root cause has been overlooked, or;
- Send a request for a footnote indicating why the report can’t be closed and need further information. Suggestions will be made for specific areas of improvement and relevant sections of the GPG that detail the requirement missed in the report may be highlighted.
The new SABRE platform and incident management database has been operational for a little over 12 months at the time of writing, first as a pilot in February 2025 and later in March 2025 for all reporters. The following data covers the reporting period for April 2025 to March 2026, so while it is recognised this does not cover the annual summary reporting period, it gives a good indication of the problems experienced with reports reviewed and assessed.
There were:
- 1267 SAE confirmation reports received that have not been excluded.
- 549 requests for footnotes due to reports not meeting the regulatory requirements cited in the GPG.
From the data, it can be concluded that out of 1267 confirmation reports there were 549 occasions (43.3%) where that report did not provide adequate information.
Learning point
- The quality of investigations and reporting should be improved to meet the requirements of Chapter 9 of the GPG.
The Good Practice Guide requirements
Everyone involved in the quality and safety of blood must be aware of the existence of the GPG and how it specifically relates to the aspects of their work involved in maintaining quality and safety. Those responsible for the design and implementation of the QMS must have a detailed knowledge of the entire guidelines. Copies of the Guide to the preparation, use and quality assurance of blood components, which contains the GPG are available to download from the Council of Europe website Guide to the preparation, use and quality assurance of blood components – European Directorate for the Quality of Medicines & HealthCare free of charge.
While the full guide is relevant, Chapter 9 covers investigation and reporting in detail. Table 30.5 highlights the most relevant sections and summary of what it covers. Full details of the reference can be found in the Guide itself.
Table 30.5: Important sections of the GPG relevant to incident management
| GPG reference | Summary | Importance |
|---|---|---|
| 9.1.4 | Adverse events should be carefully investigated for causative factors and followed up with corrective actions to prevent recurrence | This applies to ALL errors, not just those reportable to MHRA. It highlights the need to prevent recurrence and not simply to improve detection of errors already made |
| 9.1.5 | CAPA should ensure that component non-conformance or quality problems are corrected and prevented | Ensures that not only errors are investigated as above, but that all non-conforming components AND the underlying quality problems that exist must be prevented |
| 9.1.6 | Deviations from procedures should be avoided, and need to be formally documented, fully recorded and investigated for systematic problems | Investigations need to investigate the systematic reasons for error and not simply the individuals involved |
| 9.2.1 | Again, specifies the need for thorough investigation for causative factors, but also indicates the importance of recall of defective components. It also explains that Competent Authorities must be kept informed according to regulatory requirements | Highlights the need for recall and for carefully investigating and introducing CAPA for the purposes of reporting to MHRA |
| 9.4.6 | An appropriate level of root cause analysis should be applied. If the root cause (RC) cannot be determined, the most likely RC must be identified. Human error must be formally justified to ensure that process, procedural or system-based errors are not overlooked | Root cause analysis (RCA) is essential and CAPA determined even if the true RC cannot be determined. Quality problems should be identified and resolved first without holding staff solely accountable |
Learning point
- If the submitted report does not meet the above, legally required, criteria it will be returned for further information
Common reporting pitfalls
Cognitive bias as a ‘root cause’
It has been noted that cognitive bias is frequently cited as the main ‘human factor’ root cause while simultaneously being used to justify an absence of CAPA, on the grounds that it reflects an unavoidable feature of normal brain function. While cognitive bias is real and does influence human behaviour, it is incorrect to treat it as an immutable root cause whose effects cannot be mitigated.
Research on inattentional blindness, notably the Simons and Chabris (1999) selective attention experiment (Simons, 2010), demonstrates that perceptual failures arise not because information is inherently invisible, but because task instructions and expectations narrowly channel attention. In this experiment, when participants are instructed solely to count basketball passes, they frequently fail to notice an unexpected gorilla that is plainly visible. When their expectations change, the same visual information is readily perceived. Individuals failed to notice the gorilla when their attention is focused on other objects or tasks, demonstrating the impact of divided attention on perception. This illustrates that performance outcomes depend critically on task design, clarity of instruction, and cognitive load rather than on unavoidable human limitation alone.
The implication for quality and safety management in healthcare is that human factors should be addressed proactively through system design. Clear, unambiguous procedures, appropriate task allocation, allowance for distractions and workload, realistic capacity planning, and training that ensures genuine task understanding can significantly reduce error. Cognitive bias, therefore, should not be accepted passively as an endpoint explanation; it should be treated as a design constraint that can be managed through effective processes, thereby supporting the prevention of error recurrence and the robustness of the quality management system. These measures are listed in the following figure.
Figure 30.4: Measures that must be undertaken to ensure blood component quality and safety
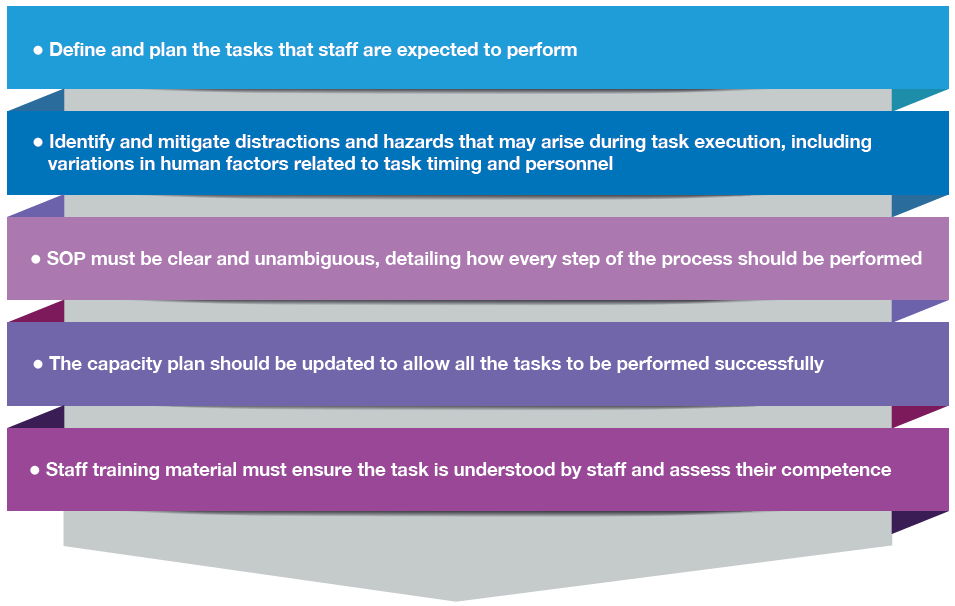
The preceding example of the gorilla experiment illustrates how the principles of the GPG can be applied to strengthen the QMS. Central to the GPG is the prevention of error recurrence. By explicitly identifying relevant human factors, organisations can address these risks through targeted system and process improvements, rather than passively accepting human error as inevitable. In practical terms, task performance improves when expectations are clearly defined: if staff are required to count basketball passes, they should be instructed accordingly; if they are expected to identify anomalies, they must be directed to do so. Where multiple objectives are required, tasks should be appropriately divided among personnel or sufficient time should be allocated to enable successful completion.
A biomedical scientist may be instructed to focus on a high‑priority task such as completing compatibility testing and verifying that the ABO and D groups match the request form. While concentrating on these checks, they may fail to notice an unexpected but equally critical anomaly, such as an expiry date on a reagent label, a subtle discrepancy in a patient identifier, or a specific transfusion requirement for the component. Like the ‘gorilla’ in the psychological experiment, the information is present and visible, but attention is directed elsewhere.
This does not indicate negligence or an inherent limitation of the individual. Rather, it reflects the effects of divided attention and cognitive load created by task design and instructions. When staff are required to perform multiple objectives simultaneously, such as speed, accuracy, vigilance for rare events, and documentation, without explicit prioritisation or sufficient time, perceptual failures become more likely.
The implication is that transfusion safety depends less on expecting individuals to ‘be more careful’ and more on designing tasks and procedures that support attention. Clear, unambiguous instructions, separation of critical checks, appropriate workload management, and system prompts for uncommon but high‑risk anomalies can significantly reduce the likelihood of missing important information. Performance outcomes, therefore, are shaped primarily by how the work is structured, rather than by unavoidable human limitation alone.
Learning point
- Cognitive bias, along with all human factors, can be mitigated in well designed and planned systems, processes and procedures.
Second checks as CAPA
It is not within the remit of MHRA to advise reporters how to design individual aspects of their QMS. If it is felt that second checks are appropriate, then they can be added into processes. However, the Haemovigilance team will review the SAE reports and assess the effectiveness of your proposed CAPA.
However, no second check has ever prevented an error occurring. A second check may have provided an additional opportunity to detect an error already made, but no second check either manual or automated has ever prevented the error that led to the SAE occurring which is being reported. The GPG directs reporters to determine process, procedural and system improvements that prevent recurrence. It does not advise that transfusion teams should only get better at detecting errors that have occurred already. As such, any mention of a second check as CAPA is not considered in the MHRA assessment of the submitted report. If there is no valid determination of RC and CAPA, the report is likely to be returned for further information. For example, in the case of a component labelling error SAE due to transposition of labels, the error is at the point of labelling, and MHRA will be looking at CAPA that directly affects the successful printing, verification and attachment of labels. Any second check that is performed occurs after the labelling has been performed. If ever successful (and many reports contain examples that the second check failed or was not even performed at all) the labelling error has not been prevented, it has only been detected.
Even in the above common and relatively simple task, a thorough and effective investigation of the human factors involved can identify system improvements. The example above describes how to potentially plan tasks and procedures, but specifically related to labelling errors.
Have all aspects of the process been considered?
- Environment
- Possible distractions
- Equipment used
- How the steps are performed including what to verify and how to verify it
- Verification of the entire donation number and not simply the ‘last 3 digits’
- Line clearance
- Is the relevant SOP clear and unambiguous?
- Have all the steps been covered? It should not be assumed that staff know what each step entails.
Do staff know how to troubleshoot problems such as printer failures and back-up procedures?
The above is not an exhaustive list but an example of the level of detailed planning that is essential in relatively simple tasks because they are a frequent and a repeated source of error.
Many errors occur because of a failure to adequately plan and describe processes in potentially weak SOP. While in many instances, there may be an SOP in place, when reviewed, it is determined to be ineffective. Examples of SOP weaknesses include:
- Many ‘SOP’ are little more than policy documents that describe an outcome to be achieved.
- Many SOP do not contain instructions of how each step is expected to be performed.
- Steps in processes are not always clear and unambiguous.
- Many will not highlight potential areas of failure, and the contingency plans expected.
Without clearly designed and unambiguous written instructions, staff are left to improvise, and improvisation leads to non-standard performance of processes and systems which can then lead to error.
Learning points
- CAPA must address the causative factors of the error made as a minimum and not just to limit the effects of the error already made.
- It should not be assumed that staff know how to perform a task.
- Tasks should be planned and regularly reviewed for effectiveness.
- Staff performance should be regularly assessed for conformance with the SOP.
Automated systems vs manual processes
Many reports determine that an error occurred because a process is manual. As a result, their CAPA is to state the introduction of a future automated process not yet planned and installed, or a simple aspiration that an automated system would address the RC. Such reports will always be returned for further investigation of RC and implementation of effective CAPA. In addition, it is important to capture the measures being taken in the short term to mitigate the risk(s) identified.
There is nothing in the BSQR or GPG that states you must introduce automated systems to replace manual systems. Both manual and automated systems are still subject to the effects of human factors. A well-designed and planned manual system is better than a badly designed and implemented automated system.
If your CAPA is to introduce an automated system based on errors involved in performing a manual one you may have potentially failed to understand the true human factors affecting performance, you will have also potentially failed to identify improvements to that manual process and your current QMS and as a consequence run the risk of repeat errors and an increased risk of harm to patients.
Human factors are still involved in the design of automated process, albeit different ones to manual processes. If we do not effectively investigate and address the RC of the current systems that led to the error being reported, we simply run the risk of introducing the possibility of different errors and failing to improve our current systems and increasing the risk of harm to patients.
Learning points
- Automated systems are not inherently better than manual processes. If the error is in the manual process CAPA must address the human factors of the manual process currently in use.
- Introduction of automated systems have their own human factors and risks involved and unless planned and well-designed may still present a risk of harm to patients.
Human factors training as CAPA
Human factors training is essential to all staff to understand how these factors can lead to human error if not adequately considered when designing our processes and systems. However, it is not a cure for an individual’s human error. Human factors training is an opportunity to review and redesign systems, processes and procedures to prevent human error.
Learning point
- Use human factors training to improve existing QMS and not as a solution to prevent human error in individuals.
Recommendations
Recommendations when presented in the report are not the same as CAPA. While it is important to recommend areas of improvement, there is no commitment to make any actual changes. The submitted report must go further and describe how these recommendations are planned to be converted into tangible and effective CAPA.
Learning point
- Convert any ‘recommendations’ in a report to CAPA.
Reminders and re-training
While it may be a good idea to remind staff of the importance of following procedures following an event, as a barrier it is ineffective. Staff should already be following those procedures, so why weren’t they? Likewise, while re-training is a good idea only if staff were not aware of the correct procedures, it is not a catch-all CAPA. One still needs to explore why a member of staff required retraining and address those factors. Retraining a member of staff does not address all the human factors that may have led to the error that occurred. In addition, it is important to look at the training regime in place to check for its effectiveness. Also, not everybody learns in the same way so potentially look at individual training needs of staff.
Learning point
- Reminders and re-training are weak barriers and do little to address the genuine human factors that led to the error being reported.
Appendices
Appendix 1: Storage subcategories
| Component expiry | A component has time expired and not been removed from the storage location according to laboratory procedures |
| Incorrect storage of component | A component has not been stored in the correct location |
| Sample expiry | A sample has expired, and the component has not been removed from the supply chain for the original patient |
| Return to stock error | A component has been returned to the supply chain in error instead of being quarantined or discarded |
| Failure to action alarm | A storage location alarm has been activated but not actioned according to the procedure |
| Storage temperature deviation | The storage temperature has gone out of specification without an alarm being activated |
| Security | A storage location is accessible to staff or public who are not authorised to do so |
| 30- or 60-minute rule | Red cells are returned to a refrigerator after 30 or 60 minutes have elapsed contrary to local procedures for return of unused red cells |
| Miscellaneous | Any other storage event affecting the quality and safety of blood or blood components |
Appendix 2: Other subcategories
| Incorrect blood component issued (IBCI) | Blood issued which does not meet the patient’s specific requirements |
| Sample processing error (SPE) | Sample incorrectly receipted into the laboratory that should have been rejected |
| Component labelling error (CLE) | Typically, transposition of labels |
| Pre-transfusion testing error (PTTE) | Any error in the process of testing patient samples and the interpretation of results |
| Component collection error (CCE) | Any error in the collection of components from storage locations, or the handover of components on collection from the laboratory |
| Data entry error (DEE) | Transcription errors of data, including both electronic and hand-written data |
| Failed recall (FR) | Failure to recall components in a timely manner |
| Unspecified (UNSPEC) | Any error affecting the quality and safety of components not specified elsewhere |
| Component available for transfusion past de-reservation (CATPD) | Expired components which were incorrectly collected, prior to their scheduled re-stock by the laboratory |
| Expired component available for transfusion (ECAT) | Any component issued for a patient, where the component expires prior to the planned transfusion |
| Incorrect blood component ordered (IBCO) | Components ordered from a blood establishment that do not meet the patient’s specific requirements |
| Handling damage (HD) | Damage to a component affecting its quality and safety |
| Incorrect blood component accepted (IBCA) | Blood accepted into a laboratory for a specific patient where the special requirements have not been matched |
Appendix 3: Human error subcategories
| Procedure performed incorrectly | Failure to carry out a step(s) correctly |
| Procedural steps omitted/wrong procedure performed | Missing a key step or not following the procedure |
| Inadequate process | Inadequate design of a process. Also includes multiple causative factors |
| Incorrect procedure | Process not properly described in the SOP |
| Ineffective training | Training not understood by operator |
| Inadequate training | Training process not fit for purpose |
| Lapsed or no training | Carrying out a procedure without any formal training |
| Inadequate QMS – staffing and workload | Staffing levels below the minimum level, or unacceptably high workload has resulted in staff making errors. It is also important to consider an appropriate skill-mix when deciding on minimum staffing levels |
| Inadequate supervision | Errors have been made by trainees or inexperienced members of staff and should have been noticed by adequate supervision |